

The Institute of Rare Diseases and the Association of Medical Students – Plovdiv with the support of the National Alliance of People with Rare Diseases and the Municipality of Plovdiv introduced the photo exhibition “Rare Nuances in The Colours of Life” today at the Roman Stadium Square at 11:00 AM.
Students from all Medical Faculties in the country, Bulgarian and foreign language studies, took part in the event. 69 authors’ photos were submitted by doctors, dentists and rehabilitators.
The current competition is held on the occasion of the International Day of Rare Diseases – 28th February – and aims to contribute to better knowledge and understanding of rare diseases to students of medical universities. As well as to increase the awareness and visibility of these diseases in the healthcare system itself and among society as a whole.
Residents of Plovdiv will have the opportunity to show their empathy for patients with rare diseases by visiting the photo exhibition in the period 20.02.2023-12.03.2023 at the Roman Stadium Square.



 Изпратете своите снимки в бланката за участие:
Изпратете своите снимки в бланката за участие: 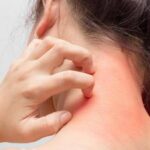 One of the modern definitions of urticaria is describing it as a
One of the modern definitions of urticaria is describing it as a