
 Синдромът на Пери-Ромберг или прогресивна хемифациална атрофия е рядко заболяване, което обикновено засяга една страна на лицето със загуба на меки и твърди тъкани. Заболяването се появява внезапно и обикновено се самоограничава след 2-10 години. Загубата на меки и твърди тъкани води до естетични и функционални дефицити, които се усложняват от наличието на други симптоми като невралгия, мигрена, епилепсия и очно засягане. Степента на деформация зависи от възрастта, в която болестта се е проявила първоначално. Колкото по-рано е началото, толкова по-тежка е деформацията. Тези пациенти претърпяват тежка психологическа травма и имат социални проблеми. Точната етиология не е известна и лечението е предимно козметично. В статията се представя доклад за три случая и преглед на литературата. Повече информация може да получите тук.
Синдромът на Пери-Ромберг или прогресивна хемифациална атрофия е рядко заболяване, което обикновено засяга една страна на лицето със загуба на меки и твърди тъкани. Заболяването се появява внезапно и обикновено се самоограничава след 2-10 години. Загубата на меки и твърди тъкани води до естетични и функционални дефицити, които се усложняват от наличието на други симптоми като невралгия, мигрена, епилепсия и очно засягане. Степента на деформация зависи от възрастта, в която болестта се е проявила първоначално. Колкото по-рано е началото, толкова по-тежка е деформацията. Тези пациенти претърпяват тежка психологическа травма и имат социални проблеми. Точната етиология не е известна и лечението е предимно козметично. В статията се представя доклад за три случая и преглед на литературата. Повече информация може да получите тук.
 Болест на Кикучи-Фуджимото (KFD) е рядка и доброкачествена причина за цервикална лимфаденопатия. Тя е анатомоклинична единица с неизвестна етиология. Потвърждението на диагнозата винаги се осигурява чрез хистологично изследване на лимфните възли. Клиничната картина понякога се бърка с лимфом или туберкулоза. Еволюцията като цяло е благоприятна със спонтанно излекуване след няколко седмици. В статията е докладван случай на 26-годишна жена, която се е консултирала за цервикална лимфаденопатия, свързана с треска. Биопсията на шийните лимфни възли потвърждава болестта на Кикучи-Фуджимото. Еволюцията е белязана от бърза регресия на лимфаденопатия при лечение с кортикостероиди. Повече информация може да получите тук.
Болест на Кикучи-Фуджимото (KFD) е рядка и доброкачествена причина за цервикална лимфаденопатия. Тя е анатомоклинична единица с неизвестна етиология. Потвърждението на диагнозата винаги се осигурява чрез хистологично изследване на лимфните възли. Клиничната картина понякога се бърка с лимфом или туберкулоза. Еволюцията като цяло е благоприятна със спонтанно излекуване след няколко седмици. В статията е докладван случай на 26-годишна жена, която се е консултирала за цервикална лимфаденопатия, свързана с треска. Биопсията на шийните лимфни възли потвърждава болестта на Кикучи-Фуджимото. Еволюцията е белязана от бърза регресия на лимфаденопатия при лечение с кортикостероиди. Повече информация може да получите тук.
 Болестта на Фабри (FD) е мултиорганно Х-свързано състояние, характеризиращо се с дефицит на лизозомния ензим алфа-галактозидаза А, което води до прогресивно интрализозомно отлагане на глоботриазозилцерамид. Целта на това изследване е да се оцени макуларната ултраструктура на съдовата мрежа, като се използва оптична кохерентна томографска ангиография (OCTA) и да се оцени макулната функция при използване на фокална електроретинография (fERG) при пациентите с болест на Фабри. Общо 20 пациента с болестта на Фабри и 17 здрави контроли участват в проучването. Във всички субекти е извършена цветна фундусна фотография, оптична кохерентна томография, OCTA и fERG. Груповите разлики са статистически оценени с различни тестове. OCTA съдови аномалии и намалени амплитуди на fERG показват субклинични признаци на микроангиопатия с ранна дисфункция на ретината при пациенти с болетта. Това проучване подчертава значението на OCTA анализ на изображения при идентифицирането на абнормна макуларна васкулатура като очен отличителен белег на FD. Повече информация може да получите тук.
Болестта на Фабри (FD) е мултиорганно Х-свързано състояние, характеризиращо се с дефицит на лизозомния ензим алфа-галактозидаза А, което води до прогресивно интрализозомно отлагане на глоботриазозилцерамид. Целта на това изследване е да се оцени макуларната ултраструктура на съдовата мрежа, като се използва оптична кохерентна томографска ангиография (OCTA) и да се оцени макулната функция при използване на фокална електроретинография (fERG) при пациентите с болест на Фабри. Общо 20 пациента с болестта на Фабри и 17 здрави контроли участват в проучването. Във всички субекти е извършена цветна фундусна фотография, оптична кохерентна томография, OCTA и fERG. Груповите разлики са статистически оценени с различни тестове. OCTA съдови аномалии и намалени амплитуди на fERG показват субклинични признаци на микроангиопатия с ранна дисфункция на ретината при пациенти с болетта. Това проучване подчертава значението на OCTA анализ на изображения при идентифицирането на абнормна макуларна васкулатура като очен отличителен белег на FD. Повече информация може да получите тук.
![]() Макар и рядко срещани, редките заболявания представляват значителен дял от въздействието на болестите в световен мащаб. Група редки генетични заболявания, наречени мукополизахаридози (MPSs), се характеризират с натрупване на частично разградени гликозаминогликани в клетките. MPS води до различни симптоми и при някои форми на заболяването до невродегенерация. Липсата на възможности за лечение на MPS с неврологично участие изисква нови възможности за терапевтично изследване. Клетъчните и генни терапии осигуряват предполагаеми алтернативи и когато са съчетани с технологии за редактиране на геноми, могат да осигурят дългосрочно лечение. Клъстерирани, внедрени кратки палиндромни повторения (CRISPR), базирани на технологии за редактиране на генома и напредъкът в изследванията за редактиране на генома, са позволили добавянето на базови редактори към репертоара на CRISPR- базирани инструменти за редактиране. Най-новите версии на базовите редактори са високоефективни по отношение на редакторите на дезоксирибонуклеинова киселина (ДНК). В този обзор се обсъжда напредъка в базовите технологии за редактиране и настоящите техники за доставяне на клетъчна и генна терапия на мястото на глобалната дегенерация при пациенти с тежки неврологични форми на MPS, на централната нервна система, включително прекъсване на кръвно-мозъчната бариера с ултразвук. Повече информация може да получите тук.
Макар и рядко срещани, редките заболявания представляват значителен дял от въздействието на болестите в световен мащаб. Група редки генетични заболявания, наречени мукополизахаридози (MPSs), се характеризират с натрупване на частично разградени гликозаминогликани в клетките. MPS води до различни симптоми и при някои форми на заболяването до невродегенерация. Липсата на възможности за лечение на MPS с неврологично участие изисква нови възможности за терапевтично изследване. Клетъчните и генни терапии осигуряват предполагаеми алтернативи и когато са съчетани с технологии за редактиране на геноми, могат да осигурят дългосрочно лечение. Клъстерирани, внедрени кратки палиндромни повторения (CRISPR), базирани на технологии за редактиране на генома и напредъкът в изследванията за редактиране на генома, са позволили добавянето на базови редактори към репертоара на CRISPR- базирани инструменти за редактиране. Най-новите версии на базовите редактори са високоефективни по отношение на редакторите на дезоксирибонуклеинова киселина (ДНК). В този обзор се обсъжда напредъка в базовите технологии за редактиране и настоящите техники за доставяне на клетъчна и генна терапия на мястото на глобалната дегенерация при пациенти с тежки неврологични форми на MPS, на централната нервна система, включително прекъсване на кръвно-мозъчната бариера с ултразвук. Повече информация може да получите тук.
 Саркоидозата (SA) не е забооляване само на белите дробове, но може да засегне и други органи, като черния дроб и далака и поради тази причина се полагат усилия за определяне на специфични критерии за изобразяване и диагностициране на всеки един орган, като основната концепция е изключването на други причини за заболяването и е важно за постигане на правилната диагноза. Ултразвукът (US) е полезен инструмент за оценка на пациенти със съмнение за коремна SA, като например при засягане на черния дроб, далака, бъбреците, панкреаса и други органи, показващи находки като органомегалия, фокални лезии и лимфаденопатия. Докато диагностицирането на коремната SA е по-предсказуемо в случай на включване на други органи (например, бели дробове), проблемът е по-сложен в случая на изолирана коремна SA. Употребата на ултразвук и ендоскопска ултразвукова еластография с контрастно усилване предостави допълнителна информация за моделите на усилване и плътността на тъканите при коремната SA. Повече информация може да получите тук.
Саркоидозата (SA) не е забооляване само на белите дробове, но може да засегне и други органи, като черния дроб и далака и поради тази причина се полагат усилия за определяне на специфични критерии за изобразяване и диагностициране на всеки един орган, като основната концепция е изключването на други причини за заболяването и е важно за постигане на правилната диагноза. Ултразвукът (US) е полезен инструмент за оценка на пациенти със съмнение за коремна SA, като например при засягане на черния дроб, далака, бъбреците, панкреаса и други органи, показващи находки като органомегалия, фокални лезии и лимфаденопатия. Докато диагностицирането на коремната SA е по-предсказуемо в случай на включване на други органи (например, бели дробове), проблемът е по-сложен в случая на изолирана коремна SA. Употребата на ултразвук и ендоскопска ултразвукова еластография с контрастно усилване предостави допълнителна информация за моделите на усилване и плътността на тъканите при коремната SA. Повече информация може да получите тук.
 Синдромът на Сатайоши е мултисистемно рядко заболяване с неизвестна етиология, въпреки че се предполага автоимунна основа. Неговите основни симптоми са: болезнени мускулни спазми, диария, алопеция и скелетни аномалии. Клиничният курс без лечение може да доведе до сериозно увреждане или смърт. Все още предстои преглед на лечението и неговия отговор. Между 1967 и 2018 г. са публикувани 64 случая на синдром на Сатайоши. Използваните лекарства могат да бъдат разделени на две основни групи на лечение: мускулни релаксанти / антиконвулсанти и кортикостероиди / имуносупресори. Дантролен подобрява мускулните симптоми при 13 от 15 случая, но не и други симптоми на заболяването. Други мускулни релаксанти или антиконвулсивни лекарствени средства показват малък или никакъв ефект. 28 от 30 случая отговарят на схема, включваща костикостероиди. Други имуносупресивни лекарства, включително циклоспорин, микофенолат мофетил, азатиоприн, метотрексат, такролимус и циклофосфамид са използвани с цел намаляване на дозата на кортикостероидите или за подобряване на ефикасността. Имуноглобулинова терапия е използвана при девет пациента и четири от тях са получили благоприятен отговор. Кортикостероидите са най-широко използваното лечение с най-добри резултати при синдрома на Сатайоши. Необходими са допълнителни проучвания, за да се определи оптималната доза и продължителността на кортикостероидния курс, както и ролята на други имуносупресори и имуноглобулинова терапия. Генетични или автоимунни маркери ще бъдат полезни за насочване на бъдещите терапии. Повече информация може да получите тук.
Синдромът на Сатайоши е мултисистемно рядко заболяване с неизвестна етиология, въпреки че се предполага автоимунна основа. Неговите основни симптоми са: болезнени мускулни спазми, диария, алопеция и скелетни аномалии. Клиничният курс без лечение може да доведе до сериозно увреждане или смърт. Все още предстои преглед на лечението и неговия отговор. Между 1967 и 2018 г. са публикувани 64 случая на синдром на Сатайоши. Използваните лекарства могат да бъдат разделени на две основни групи на лечение: мускулни релаксанти / антиконвулсанти и кортикостероиди / имуносупресори. Дантролен подобрява мускулните симптоми при 13 от 15 случая, но не и други симптоми на заболяването. Други мускулни релаксанти или антиконвулсивни лекарствени средства показват малък или никакъв ефект. 28 от 30 случая отговарят на схема, включваща костикостероиди. Други имуносупресивни лекарства, включително циклоспорин, микофенолат мофетил, азатиоприн, метотрексат, такролимус и циклофосфамид са използвани с цел намаляване на дозата на кортикостероидите или за подобряване на ефикасността. Имуноглобулинова терапия е използвана при девет пациента и четири от тях са получили благоприятен отговор. Кортикостероидите са най-широко използваното лечение с най-добри резултати при синдрома на Сатайоши. Необходими са допълнителни проучвания, за да се определи оптималната доза и продължителността на кортикостероидния курс, както и ролята на други имуносупресори и имуноглобулинова терапия. Генетични или автоимунни маркери ще бъдат полезни за насочване на бъдещите терапии. Повече информация може да получите тук.
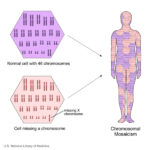 Синдромът на Търнър е рядко състояние при жените, което е свързано или с пълна, или с частична загуба на една Х-хромозома, често в мозаечни кариотипове. Синдромът на Търнър се свързва с нисък ръст, забавен пубертет, яйчникова дисгенезия, хипергонадотропен хипогонадизъм, безплодие, вродени малформации на сърцето, ендокринни заболявания като захарен диабет тип 1 и тип 2, остеопороза и автоимунни заболявания. Заболеваемостта и смъртността са по-големи при жени със синдром на Търнър в сравнение с общото население и засягането на множество органи и системи през всички етапи от живота изисква мултидисциплинарен подход в лечението. Въпреки характерния фенотип, диагностичното забавяне може да бъде значително и средната възраст за поставяне на диагнозата е около 15-годишна възраст. Въпреки това, са постигнати много важни клинични постижения, обхващащи всички специалности, включени в грижата за момичета и жени със синдром на Търнър. Статията представя актуализиран преглед на синдрома на Търнър, който обхваща напредъка в генетичните и геномни механизми на заболяването, свързаните с него нарушения и мултидисциплинарните подходи в лечението на пациентите, включително и терапия с растежен хормон и хормонозаместителна терапия. Повече информация може да получите тук.
Синдромът на Търнър е рядко състояние при жените, което е свързано или с пълна, или с частична загуба на една Х-хромозома, често в мозаечни кариотипове. Синдромът на Търнър се свързва с нисък ръст, забавен пубертет, яйчникова дисгенезия, хипергонадотропен хипогонадизъм, безплодие, вродени малформации на сърцето, ендокринни заболявания като захарен диабет тип 1 и тип 2, остеопороза и автоимунни заболявания. Заболеваемостта и смъртността са по-големи при жени със синдром на Търнър в сравнение с общото население и засягането на множество органи и системи през всички етапи от живота изисква мултидисциплинарен подход в лечението. Въпреки характерния фенотип, диагностичното забавяне може да бъде значително и средната възраст за поставяне на диагнозата е около 15-годишна възраст. Въпреки това, са постигнати много важни клинични постижения, обхващащи всички специалности, включени в грижата за момичета и жени със синдром на Търнър. Статията представя актуализиран преглед на синдрома на Търнър, който обхваща напредъка в генетичните и геномни механизми на заболяването, свързаните с него нарушения и мултидисциплинарните подходи в лечението на пациентите, включително и терапия с растежен хормон и хормонозаместителна терапия. Повече информация може да получите тук.
 Анемията на Diamond-Blackfan е рядка вродена аплазия на червените кръвни клетки, характеризираща се с неуспешна еритропоеза, вродени аномалии в до 50% от пациентите, забавяне на растежа в до 30% от пациентите и предразположеност към злокачествени заболявания. Заболяването е както клинично, така и генетично хетерогенно състояние, вариращо от фини асимптоматични еритроидни аномалии до неимунен фетален хидропс. Съвременните възможности за лечение включват кортикостероидна терапия, хронични трансфузии на червени кръвни клетки и трансплантация на хемопоетични стволови клетки с генна терапия. В публикацията се съобщава за първия документиран случай на анемията на Diamond-Blackfan в Южна Африка при едно европеидно момиче с вторична хетерозиготна делеция на ген в RPL35A. Ограничените ресурси, липсата на тестове, непознаването на редките заболявания, разширената диференциална диагноза и свързаната неутропения са довели до забавяне в диагностиката на заболяването. Този случай напомня на клиничните специалисти, че това заболяване може да доведе и до апластична анемия и подчертава трудностите и пречките при диагностицирането му в ограничените от ресурси страни. Случаят напомня още, че може да е причина за анемия при бебетата, за ограниченията при поставянето на диагноза в здравните системи с недостатъчни ресурси и необходимостта от стандартизирани протоколи за лечение, приложими за страните с ограничен ресурс. Повече информация може да получите тук.
Анемията на Diamond-Blackfan е рядка вродена аплазия на червените кръвни клетки, характеризираща се с неуспешна еритропоеза, вродени аномалии в до 50% от пациентите, забавяне на растежа в до 30% от пациентите и предразположеност към злокачествени заболявания. Заболяването е както клинично, така и генетично хетерогенно състояние, вариращо от фини асимптоматични еритроидни аномалии до неимунен фетален хидропс. Съвременните възможности за лечение включват кортикостероидна терапия, хронични трансфузии на червени кръвни клетки и трансплантация на хемопоетични стволови клетки с генна терапия. В публикацията се съобщава за първия документиран случай на анемията на Diamond-Blackfan в Южна Африка при едно европеидно момиче с вторична хетерозиготна делеция на ген в RPL35A. Ограничените ресурси, липсата на тестове, непознаването на редките заболявания, разширената диференциална диагноза и свързаната неутропения са довели до забавяне в диагностиката на заболяването. Този случай напомня на клиничните специалисти, че това заболяване може да доведе и до апластична анемия и подчертава трудностите и пречките при диагностицирането му в ограничените от ресурси страни. Случаят напомня още, че може да е причина за анемия при бебетата, за ограниченията при поставянето на диагноза в здравните системи с недостатъчни ресурси и необходимостта от стандартизирани протоколи за лечение, приложими за страните с ограничен ресурс. Повече информация може да получите тук.
 Хемофилия А (НА) е Х-свързано наследствено нарушение на кръвта, причинено от дефицит на коагулационен фактор VIII (FVIII). Едно от най-големите усложнения при лечението на НА е развитието на неутрализиращи алоантитела, известни като инхибитори на FVIII. Пациентите, страдащи от HA, при които се образуват инхибитори на FVIII, имат ограничени възможности за лечение и срещат по-големи проблеми, свързани с лечението и заболяването, отколкото пациентите на HA без FVIII инхибитори. Емицизумаб, наскоро одобрено биспецифично моноклонално антитяло, имитира функцията на FVIIIa чрез свързване на FIXa и FX за възстановяване на ефективната хемостаза. В публикацията се обсъждат текущите лабораторни методи за мониторинг, включително активирано парциално тромбопластиново време, FVIII едноетапни анализи на кръвосъсирването, FVIII хромогенни анализи и глобални анализи на коагулациите; посочва се защо тези конвенционални методи могат да бъдат неподходящи за наблюдение на пациенти с НА, получаващи емицизумаб и се предлагат алтернативни методи, приложими за мониториране на лечението на НА в един развиващ се пейзаж. Повече информация може да получите тук.
Хемофилия А (НА) е Х-свързано наследствено нарушение на кръвта, причинено от дефицит на коагулационен фактор VIII (FVIII). Едно от най-големите усложнения при лечението на НА е развитието на неутрализиращи алоантитела, известни като инхибитори на FVIII. Пациентите, страдащи от HA, при които се образуват инхибитори на FVIII, имат ограничени възможности за лечение и срещат по-големи проблеми, свързани с лечението и заболяването, отколкото пациентите на HA без FVIII инхибитори. Емицизумаб, наскоро одобрено биспецифично моноклонално антитяло, имитира функцията на FVIIIa чрез свързване на FIXa и FX за възстановяване на ефективната хемостаза. В публикацията се обсъждат текущите лабораторни методи за мониторинг, включително активирано парциално тромбопластиново време, FVIII едноетапни анализи на кръвосъсирването, FVIII хромогенни анализи и глобални анализи на коагулациите; посочва се защо тези конвенционални методи могат да бъдат неподходящи за наблюдение на пациенти с НА, получаващи емицизумаб и се предлагат алтернативни методи, приложими за мониториране на лечението на НА в един развиващ се пейзаж. Повече информация може да получите тук.
 Редките очни патологии имат важно влияние върху качеството на живот на пациентите, тъй като често увреждането е двустранно и въпреки, че са асиметрични причиняват значително намаляване на зрителната острота. Често могат да протекат асимптоматично до сравнително късен етап и поради това диагнозата често се забавя. Разбирането на патофизиологията, диагнозата и лечението на заболяването може да помогне на лекарите от първичната медицинска помощ да насочат пациентите с висок риск за цялостно офталмологично изследване и за по-активно участие в грижите за себе си. Голям процент от тези редки болести нямат лечение, което да е одобрено от FDA. Изследването и мониторинга на пациенти с редки офталмологични нарушения представлява ключов компонент в проекта на Университетската спешна болница, Букурещ, Румъния – Клиника по офталмология. Регистрите с редки болести са водещи инструменти за разработване на клинични изследвания за редки заболявания, подобряване на достъпа на пациентите до нови диагностични методи, проследяване и нови терапии. Към настоящия момент европейският списък на редките заболявания включва 53 офталмологични заболявания, които са класифицирани като редки болести и други 103 системни заболявания с офталмологично засягане, от общо 7000 редки заболявания. Повече информация може да получите тук.
Редките очни патологии имат важно влияние върху качеството на живот на пациентите, тъй като често увреждането е двустранно и въпреки, че са асиметрични причиняват значително намаляване на зрителната острота. Често могат да протекат асимптоматично до сравнително късен етап и поради това диагнозата често се забавя. Разбирането на патофизиологията, диагнозата и лечението на заболяването може да помогне на лекарите от първичната медицинска помощ да насочат пациентите с висок риск за цялостно офталмологично изследване и за по-активно участие в грижите за себе си. Голям процент от тези редки болести нямат лечение, което да е одобрено от FDA. Изследването и мониторинга на пациенти с редки офталмологични нарушения представлява ключов компонент в проекта на Университетската спешна болница, Букурещ, Румъния – Клиника по офталмология. Регистрите с редки болести са водещи инструменти за разработване на клинични изследвания за редки заболявания, подобряване на достъпа на пациентите до нови диагностични методи, проследяване и нови терапии. Към настоящия момент европейският списък на редките заболявания включва 53 офталмологични заболявания, които са класифицирани като редки болести и други 103 системни заболявания с офталмологично засягане, от общо 7000 редки заболявания. Повече информация може да получите тук.