
 Ralitza Raycheva, Kostadin Kostadinov, Georgi Iskrov, Georgi Stefanov, Elena Mitova and Rumen Stefanov, as part of the Institute for Rare Diseases and the Department of Social Medicine and Public Health at the Medical University – Plovdiv, actively participate in the Screen4Care project, which started in 2021. Screen4Care offers an innovative research approach to accelerate rare disease diagnosis, which is based on two central pillars: genetic newborn screening and digital technologies.
Ralitza Raycheva, Kostadin Kostadinov, Georgi Iskrov, Georgi Stefanov, Elena Mitova and Rumen Stefanov, as part of the Institute for Rare Diseases and the Department of Social Medicine and Public Health at the Medical University – Plovdiv, actively participate in the Screen4Care project, which started in 2021. Screen4Care offers an innovative research approach to accelerate rare disease diagnosis, which is based on two central pillars: genetic newborn screening and digital technologies.
Current research study “Landscape analysis of available European data sources amenable for machine learning and recommendations on usability for rare diseases screening” has been executed by Georgi Iskrov, Ralitsa Raicheva, Kostadin Kostadinov, Georgi Stefanov, Elena Mitova and Rumen Stefanov from BAPES, Institute for Rare Diseases team together with Merja Vakevainen, Kaisa Elomaa, Yuen-Sum Man, Edith Gross, Jana Zschüntzsch, Richard Röttger and is published in the prestigious international journal “Orphanet Journal of Rare Diseases” (Impact factor: 3.7).
Patient registries and databases are pivotal tools in advancing clinical research for rare diseases, as well as in improving patient care and healthcare planning. This study aims to conduct a comprehensive analysis of available European data sources suitable for machine learning (ML) in rare diseases screening, with a focus on their adherence to the FAIR (findable, accessible, interoperable, reusable) principles, legal compliance, and business considerations.
Between March 2022 and December 2022, a cross-sectional study was conducted using a semi-structured questionnaire distributed to database contacts. Drawing from relevant scientific literature, quantitative and qualitative research, and scoping reviews, the questionnaire was designed to capture insights into challenges in mapping European rare disease databases. Bayesian models were employed to assess database characteristics associated with FAIR adherence, legal compliance, and business considerations.
Analysis of 330 unique replies revealed diverse database characteristics and geographical scopes. Notably, European registries exhibited the highest overall FAIR adherence, while registries with regional and “other” geographical scopes ranked lowest in FAIR adherence. Despite the majority of databases being active and established after 2000, there remains a significant reluctance among EU health databases to share patient information. This reluctance is often attributed to legal barriers and concerns about data privacy. Only a small proportion of respondents expressed a direct willingness to contribute their databases to initiatives like the Screen4Care project.
The most important results of this study demonstrate not enough sufficient FAIR principles adherence and low willingness of the EU health databases to share patient information, combined with some legislation incapacities, resulting in barriers to the secondary use of data. Read the full article here.
Screen4Care project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under Grant Agreement No. 101034427. The JU receives support from the European Union’s Horizon 2020 research and innovation programme and EFPIA.

 Newborn screening plays a crucial role in early detection of conditions in newborns. This leads to timely therapeutic interventions that can significantly improve outcomes. Ensuring quality and standardization of newborn screening procedures is crucial for its effectiveness. We had the privilege to interview Peter Schielen, Director of the International Society for Neonatal Screening (ISNS) office, an expert in quality issues and standardization of neonatal screening, and Editor-in-Chiefof the scientific journal “International Journal of Neonatal Screening”. Peter Schielen is a guest lecturer at the 10th International Symposium on Health Technology Assessment (5.04.2024 in Sofia) by invitation of the Institute for Rare Diseases.
Newborn screening plays a crucial role in early detection of conditions in newborns. This leads to timely therapeutic interventions that can significantly improve outcomes. Ensuring quality and standardization of newborn screening procedures is crucial for its effectiveness. We had the privilege to interview Peter Schielen, Director of the International Society for Neonatal Screening (ISNS) office, an expert in quality issues and standardization of neonatal screening, and Editor-in-Chiefof the scientific journal “International Journal of Neonatal Screening”. Peter Schielen is a guest lecturer at the 10th International Symposium on Health Technology Assessment (5.04.2024 in Sofia) by invitation of the Institute for Rare Diseases. 10th INTERNATIONAL SYMPOSIUM
10th INTERNATIONAL SYMPOSIUM The integration of whole genome sequencing into all aspects of modern medicine represents the next step in the evolution of healthcare. Using this technology, scientists and physicians can observe the entire human genome comprehensively, generating a plethora of new sequencing data. Modern computational analysis entails advanced algorithms for variant detection, as well as complex models for classification. Data science and machine learning play a crucial role in the processing and interpretation of results, using enormous databases and statistics to discover new and support current genotype-phenotype correlations.
The integration of whole genome sequencing into all aspects of modern medicine represents the next step in the evolution of healthcare. Using this technology, scientists and physicians can observe the entire human genome comprehensively, generating a plethora of new sequencing data. Modern computational analysis entails advanced algorithms for variant detection, as well as complex models for classification. Data science and machine learning play a crucial role in the processing and interpretation of results, using enormous databases and statistics to discover new and support current genotype-phenotype correlations.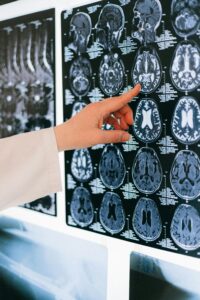 Benign tumors, called vestibular schwannomas, originate from the eighth cranial nerve and have a variable natural history. Clinically and radiologically, the solid variety is distinguished from cystic vestibular schwannomas (VS) forms, a rare benign subgroup.
Benign tumors, called vestibular schwannomas, originate from the eighth cranial nerve and have a variable natural history. Clinically and radiologically, the solid variety is distinguished from cystic vestibular schwannomas (VS) forms, a rare benign subgroup. Silver-Russell syndrome (SRS) is a rare disease with an incidence of 1:30,000 to 1:100,000 newborns. Due to apparent clinical heterogeneity, a large proportion of patients with SRS are undiagnosed. The main characteristics of the syndrome are intrauterine retardation and postnatal growth retardation, dysmorphic stigmas, and malformations of other organs and systems.
Silver-Russell syndrome (SRS) is a rare disease with an incidence of 1:30,000 to 1:100,000 newborns. Due to apparent clinical heterogeneity, a large proportion of patients with SRS are undiagnosed. The main characteristics of the syndrome are intrauterine retardation and postnatal growth retardation, dysmorphic stigmas, and malformations of other organs and systems.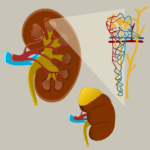 IgA nephropathy (ORPHA code: 34145) is a rare, serious condition in the complex field of renal diseases. It is an immune-mediated kidney disease that progresses over time. It emerges as a significant contributor to renal failure on a global scale. This article provides new information about this complex renal disorder. It was co-authored by Elena Mitova, MD and Georgi Stefanov, MD from the Institute for Rare Diseases in Plovdiv, and prof. Julian Ananiev of department of General and Clinical Pathology, Forensic Medicine, Deontology and Dermatovenerology, Medical Faculty, Trakia University, Stara Zagora. Their combined efforts create a narrative that clarifies the complex epidemiological environment of IgA nephropathy.
IgA nephropathy (ORPHA code: 34145) is a rare, serious condition in the complex field of renal diseases. It is an immune-mediated kidney disease that progresses over time. It emerges as a significant contributor to renal failure on a global scale. This article provides new information about this complex renal disorder. It was co-authored by Elena Mitova, MD and Georgi Stefanov, MD from the Institute for Rare Diseases in Plovdiv, and prof. Julian Ananiev of department of General and Clinical Pathology, Forensic Medicine, Deontology and Dermatovenerology, Medical Faculty, Trakia University, Stara Zagora. Their combined efforts create a narrative that clarifies the complex epidemiological environment of IgA nephropathy. Paroxysmal nocturnal hemoglobinuria (PNH) is a chronic multisystem, progressive and life-threatening disease which is characterized by intravascular hemolysis, thrombotic complications, severe infections and bone marrow failure. PNH is a rare disease with estimated prevalence of 2/100 000 by Orphanet data.
Paroxysmal nocturnal hemoglobinuria (PNH) is a chronic multisystem, progressive and life-threatening disease which is characterized by intravascular hemolysis, thrombotic complications, severe infections and bone marrow failure. PNH is a rare disease with estimated prevalence of 2/100 000 by Orphanet data. NESCAV syndrome (Neurodegeneration and spasticity with or without cerebellar atrophy or cortical visual impair-ment) is an autosomal dominant neurodegenerative disorder char-acterized by developmental delay, progressive spasticity and behav-ioural abnormalities. Additional features may include cortical visual impairment, often associated with optic atrophy, axonal peripheral neuropathy, seizures, dysautonomia, ataxia, and dystonia.
NESCAV syndrome (Neurodegeneration and spasticity with or without cerebellar atrophy or cortical visual impair-ment) is an autosomal dominant neurodegenerative disorder char-acterized by developmental delay, progressive spasticity and behav-ioural abnormalities. Additional features may include cortical visual impairment, often associated with optic atrophy, axonal peripheral neuropathy, seizures, dysautonomia, ataxia, and dystonia. The execution of clinical trials for rare diseases continues to face methodological challenges. The variable and often very small number of patients afflicted by these conditions exacerbates statistical difficulties, which unfortunately have received inadequate attention. This paper introduces the iSTORE project, shedding light on its objectives and methodological strategies.
The execution of clinical trials for rare diseases continues to face methodological challenges. The variable and often very small number of patients afflicted by these conditions exacerbates statistical difficulties, which unfortunately have received inadequate attention. This paper introduces the iSTORE project, shedding light on its objectives and methodological strategies.