
 За 15-та поредна година предстои да се проведе Национална конференция за редки болести и лекарства сираци, организирана от Институт по редки болести.
За 15-та поредна година предстои да се проведе Национална конференция за редки болести и лекарства сираци, организирана от Институт по редки болести.
Конференцията е най-значимият форум в областта на редките болести в България, който се провежда от 2010 г. насам. Събитието традиционно обединява всички заинтересовани страни – медицински специалисти, пациенти, студенти по медицина, здравни власти и представители на индустрията, за да представят и обсъдят новостите в диагностиката, лечението и проследяването на редките болести и достъпа до иновации.
Тази година по-време на събитието ще се отбележат 20 години от създаването на Институт по редки болести, чийто екип успешно е осъществил развитието на националната политика и стратегия за редки болести, предоставил е информация и консултации за пациенти и медицински специалисти, както и успешно е реализирал редица обучения, проекти и семинари.
Място: хотел Империал Пловдив гр. Пловдив
Краен срок за ранна регистрация: 1 МАЙ 2024
Краен срок за късна регистрация: 1 СЕПТЕМВРИ 2024
Краен срок за изпращане на резюмета: 1 август 2024 г.
За контакти с организационния комитет: congress@raredis.org
Допълнителнa информация можете да намерите на: XV НАЦИОНАЛНА КОНФЕРЕНЦИЯ ЗА РЕДКИ БОЛЕСТИ И ЛЕКАРСТВА СИРАЦИ – Виртуален Конгресен Център (raredis.org)

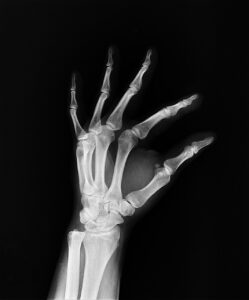 Това изследване има за цел да опише голяма група от италиански пациенти, засегнати от остеогенезис имперфекта, предоставяйки картина на клиничните костни и извънкостни характеристики и молекулярния фон, за да се подобрят познанията за заболяването и да се избере подходящо лечение в клиничната практика.
Това изследване има за цел да опише голяма група от италиански пациенти, засегнати от остеогенезис имперфекта, предоставяйки картина на клиничните костни и извънкостни характеристики и молекулярния фон, за да се подобрят познанията за заболяването и да се избере подходящо лечение в клиничната практика. В страните от Европейския съюз заболяване, което засяга по-малко от 5 души на 10 000, се счита за рядко. Поради недостига на опит и сложността на редките болести, пациентите могат да останат без диагноза години наред. Положени са усилия за включване на ORPHA – кодовете в здравно-информационните системи, което позволява кодирането на редки болести. Преди това тази номенклатура обхващаше само специфични редки заболявания, оставяйки тези с недиагностицирани състояния без кодове. Признаването на статут на редки болести е от решаващо значение за пациентите, подпомагайки възстановяването на разходите за грижи, осигурявайки социална и психологическа подкрепа и предоставяйки достъп до научни постижения, дори без точна диагноза.
В страните от Европейския съюз заболяване, което засяга по-малко от 5 души на 10 000, се счита за рядко. Поради недостига на опит и сложността на редките болести, пациентите могат да останат без диагноза години наред. Положени са усилия за включване на ORPHA – кодовете в здравно-информационните системи, което позволява кодирането на редки болести. Преди това тази номенклатура обхващаше само специфични редки заболявания, оставяйки тези с недиагностицирани състояния без кодове. Признаването на статут на редки болести е от решаващо значение за пациентите, подпомагайки възстановяването на разходите за грижи, осигурявайки социална и психологическа подкрепа и предоставяйки достъп до научни постижения, дори без точна диагноза. Това проучване включва над 11.6 милиона скринирани новородени за болестта на Помпе от 29 различни универсални програми за скрининг в 8 страни и 4 континента. Болестността на болестта на Помпе при раждане е 1:18,711, без доказателства за разлики между популации от европейски, латиноамерикански или азиатски произход, въпреки че могат да съществуват разлики за подтипове на болестта на Помпе. Това проучване също така сравнява тези резултати, базирани на директно откриване на заболяването и анализирани с помощта на биномен метод заедно с анализ на мощността, с други методи за оценка на ‘честотата’ на редките генетични заболявания.
Това проучване включва над 11.6 милиона скринирани новородени за болестта на Помпе от 29 различни универсални програми за скрининг в 8 страни и 4 континента. Болестността на болестта на Помпе при раждане е 1:18,711, без доказателства за разлики между популации от европейски, латиноамерикански или азиатски произход, въпреки че могат да съществуват разлики за подтипове на болестта на Помпе. Това проучване също така сравнява тези резултати, базирани на директно откриване на заболяването и анализирани с помощта на биномен метод заедно с анализ на мощността, с други методи за оценка на ‘честотата’ на редките генетични заболявания. Редките болести, като наследствените метаболитни заболявания, бяха идентифицирани като здравен приоритет в рамките на Европейския съюз преди повече от 20 години и станаха неразделна част от здравните програми на ЕС и европейските референтни мрежи. Притежавайки потенциала за обединяване на данни, за постигане на достатъчен размер на извадката, за преодоляване на празнините в знанията за редките заболявания и за насърчаване на епидемиологични и клинични изследвания, регистрите на пациентите са признати за ключови инструменти за основана на доказателства медицина за лица с редки заболявания.
Редките болести, като наследствените метаболитни заболявания, бяха идентифицирани като здравен приоритет в рамките на Европейския съюз преди повече от 20 години и станаха неразделна част от здравните програми на ЕС и европейските референтни мрежи. Притежавайки потенциала за обединяване на данни, за постигане на достатъчен размер на извадката, за преодоляване на празнините в знанията за редките заболявания и за насърчаване на епидемиологични и клинични изследвания, регистрите на пациентите са признати за ключови инструменти за основана на доказателства медицина за лица с редки заболявания. Ангажираността на пациентите се определя като значимо участие и активно партньорство на пациентите и ключовите партньори по време на целия изследователски проект. Тази статия разглежда значението на разработването на план за ангажиране на пациентите за насърчаване на по-доброто съгласуване на научните изследвания с реалните нужди и притеснения на пациентите и клиницистите.
Ангажираността на пациентите се определя като значимо участие и активно партньорство на пациентите и ключовите партньори по време на целия изследователски проект. Тази статия разглежда значението на разработването на план за ангажиране на пациентите за насърчаване на по-доброто съгласуване на научните изследвания с реалните нужди и притеснения на пациентите и клиницистите. Хората, живеещи с редки болести, и техните семейства все още имат неудовлетворени нужди, включително дълго време за поставяне на диагноза, ограничени възможности за лечение и огромно психосоциално бреме поради липсата на координирани, интегрирани грижи. Постигането на универсално здравно покритие за редки болести зависи от фундаментални здравни детерминанти и необходимост от различни решения. Това включва консолидиране на експертен опит чрез високотехнологични експертни центрове, установяване на ефективен път за получаване на здравни грижи, насърчаване на широко сътрудничество на европейско и световно ниво в научните изследвания и здравеопазването и поставяне на пациентите в центъра на грижите.
Хората, живеещи с редки болести, и техните семейства все още имат неудовлетворени нужди, включително дълго време за поставяне на диагноза, ограничени възможности за лечение и огромно психосоциално бреме поради липсата на координирани, интегрирани грижи. Постигането на универсално здравно покритие за редки болести зависи от фундаментални здравни детерминанти и необходимост от различни решения. Това включва консолидиране на експертен опит чрез високотехнологични експертни центрове, установяване на ефективен път за получаване на здравни грижи, насърчаване на широко сътрудничество на европейско и световно ниво в научните изследвания и здравеопазването и поставяне на пациентите в центъра на грижите.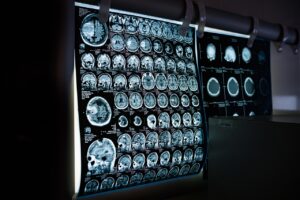 Саркоидозата се дефинира като имуномедиирано мултиорганно грануломатозно заболяване с неизвестна етиология, което се характеризира с наличието на множество неказеозни грануломи при липса на определена инфекциозна или токсична причина. Невросаркоидоза (НС) възниква, когато саркоидните грануломи засягат централната или периферната нервна система. Саркоидозата обикновено се проявява с неспецифични прояви, включително суха кашлица, умора, нощно изпотяване, загуба на тегло, кожни промени и очни прояви. Много пациенти, които развиват НС, имат неврологични прояви в рамките на две години след диагностицирането на саркоидоза. В тази статия се представя случай на новодиагностицирана саркоидоза при 49-годишен пациент от мъжки пол, първоначално с неврологични прояви с неизвестен произход, по-късно идентифицирани като НС при биопсия на периферни лимфни възли с неказеозиращ гранулом. Прочетете цялата статия
Саркоидозата се дефинира като имуномедиирано мултиорганно грануломатозно заболяване с неизвестна етиология, което се характеризира с наличието на множество неказеозни грануломи при липса на определена инфекциозна или токсична причина. Невросаркоидоза (НС) възниква, когато саркоидните грануломи засягат централната или периферната нервна система. Саркоидозата обикновено се проявява с неспецифични прояви, включително суха кашлица, умора, нощно изпотяване, загуба на тегло, кожни промени и очни прояви. Много пациенти, които развиват НС, имат неврологични прояви в рамките на две години след диагностицирането на саркоидоза. В тази статия се представя случай на новодиагностицирана саркоидоза при 49-годишен пациент от мъжки пол, първоначално с неврологични прояви с неизвестен произход, по-късно идентифицирани като НС при биопсия на периферни лимфни възли с неказеозиращ гранулом. Прочетете цялата статия  Регистрите за редки болести (РРБ) улесняват наблюдението на редките болести чрез обединяване на малки набори от данни, за да се повишат клиничните и епидемиологични познания за редките болести и да се насърчат най-добрите практики, ориентирани към пациента. Целта на това проучване е да разбере текущото състояние на РРБ в Австралия, събраните данни, въздействието върху резултатите на пациентите, моделите на финансиране и бариерите и факторите, които позволяват тяхното създаване и поддържане.
Регистрите за редки болести (РРБ) улесняват наблюдението на редките болести чрез обединяване на малки набори от данни, за да се повишат клиничните и епидемиологични познания за редките болести и да се насърчат най-добрите практики, ориентирани към пациента. Целта на това проучване е да разбере текущото състояние на РРБ в Австралия, събраните данни, въздействието върху резултатите на пациентите, моделите на финансиране и бариерите и факторите, които позволяват тяхното създаване и поддържане.